
A guide to help investigators understand the critical regulatory requirements for the informed consent process, from drafting your consent materials to administering informed consent with research participants. This resource provides guidance on special considerations needed for the NH population including how to collect consent when there are cognitive concerns.

This guide is intended for investigators and study teams planning or conducting research in nursing homes (NHs) and other long-term care settings, as well as NH partners supporting study implementation. It summarizes practical, ethically grounded approaches to informed consent in this setting, including when consent waivers may be appropriate, how to assess capacity and obtain resident assent, how to identify legally authorized representatives (LARs), and what to document. This guide is compliant with Federal Regulations governing the conduct of research (45 CRF 46.101b).
This guide focuses on informed consent for studies involving NH residents, but research studies that involve NH staff or resident family members as participants may also require informed consent. The tables and suggested workflows included below are helpful reference tools to use when designing your protocol and preparing Institutional Review Board (IRB) materials.
Why Informed Consent Requires Special Consideration in NH Research
NH residents as a vulnerable population. NH residents are a vulnerable population under Federal Guidelines because of their dependence on others for basic care. Some residents lack capacity to provide informed consent for research due to cognitive impairment. Still others have hearing or vision impairments that impact their ability to participate in informed consent discussions. Designing an informed consent process that is not coercive, involves the right decision makers, and accommodates impairments is critical.
Impact of low consent rates on research findings. While community-dwelling adults agree to participate in research at similar rates as younger adults (65%)(1), participation rates in NHs are often substantially lower with some studies enrolling fewer than one-third of eligible individuals. Low participation rates exacerbate “healthy volunteer bias” in which individuals consenting for research are healthier or more motivated than those who do not. Healthy volunteer bias results in research findings that do not generalize to the broader NH population and may underestimate intervention risk and/or overestimate its benefits. Designing an informed consent process that supports recruiting and retaining a representative sample of NH residents can reduce this bias. These factors make it essential that investigators design consent processes that both protect residents and enable representative participation.
Consent Waivers
Appropriate use of waivers for all or part of the informed consent process reduces the burden on research participants and can increase participation rates and generalizability. Common NH study types and their potential use of waivers are summarized below and in Table 1.
Full Waiver of Consent. Full waivers are most frequently used for retrospective chart reviews, secondary data analysis, or use of archived specimens where no new data are collected. However, even randomized trials may qualify for a full waiver if outcomes are assessed using data collected in routine clinical care, compare accepted treatments or randomize minimal risk interventions at a group level. Full waivers of consent can only be granted for studies that pose minimal risk to participants, do not negatively impact their rights or welfare, and could not practicably be completed without the waiver. If collecting individually identifiable data (including dates), investigators must also show that it is not possible to conduct the study without identifiers.
Even if a full waiver of consent is granted, it may still be appropriate to inform residents and/or families about the ongoing study and offer them the opportunity to opt out. State regulators’ expectations about participation in research vary, and investigators should discuss the issue of resident notification with the NH leadership. One strategy is to provide a letter that the NH can distribute to patients and families/LARs, describing how they can decline to participate (e.g., calling a phone number, notifying a NH representative). Whenever appropriate, providing residents or families/LARs with the results of the study at its conclusion is also required; options include investigator presentations at resident or family council meetings or mailings to residents and proxies.
Waiver of Documentation of Consent. In some situations where individual informed consent is needed, it may be possible to obtain a waiver of documentation. Under this waiver, participants receive all the required information about the study but verbally or implicitly provide consent (e.g., by participating in an interview or completing a questionnaire) rather than signing a consent form. This option is particularly useful in NH research because many potential participants have visual or motor impairments that make it difficult to sign a form. It is also helpful to request a waiver of documentation for studies that anticipate obtaining consent from families/LARs over the phone to minimize the burden of asking them to provide a signed consent form. A waiver of documentation can only be granted for minimal risk studies, or in studies involving sensitive data in which the signed form is the only link to the participant.
IRBs typically require investigators to submit scripts reflecting the information that will be verbally provided to potential participants. The script must address all the required elements of informed consent (see table 4). Some IRBs will also require that participants be given a written study description with investigator contact information.
Multiple Consent Waivers. Studies may employ multiple types of consent waivers for different aspects of data collection. For example, a study might request a full waiver of consent for collecting outcome data from the Minimum Data Set (MDS), plus a waiver of documentation for residents and proxies to complete patient reported outcomes surveys.
Table 1: Potential for Waivers of the Informed Consent Process by Study Type
| Study Design | Examples | Potential Waiver Options |
|---|---|---|
| FDA registration studies | Drug, device or vaccine trial | None |
| Individually randomized trials | Novel resident interventions, medications for off label uses, comparative effectiveness trials | Greater than Minimal Risk – None Minimal Risk New Intervention – Waiver of Documentation for residents with visual/motor impairment and/or telephone consent from Legally Authorized Representative (LAR) Comparative Effectiveness of Established Treatments – Full Waiver if using routinely collected outcome data; Waiver of Documentation if collecting new data |
| Group randomized trials | Infection control interventions, EHR alerts, staff education programs | Greater than Minimal Risk – None Minimal Risk – Full Waiver if using routinely collected outcome data; Waiver of Documentation if collecting new data |
| Prospective observational studies | Evaluation of quality improvement initiatives, health services interventions, new payment models | Routinely collected data – Full Waiver Collecting new data – Waiver of Documentation |
| Retrospective studies | Epidemiologic studies, Target emulation trials using existing data | Full Waiver |
| Cross-sectional studies | Qualitative interviews, surveys | Waiver of Documentation |
Figure 1: When Informed Consent Is Required
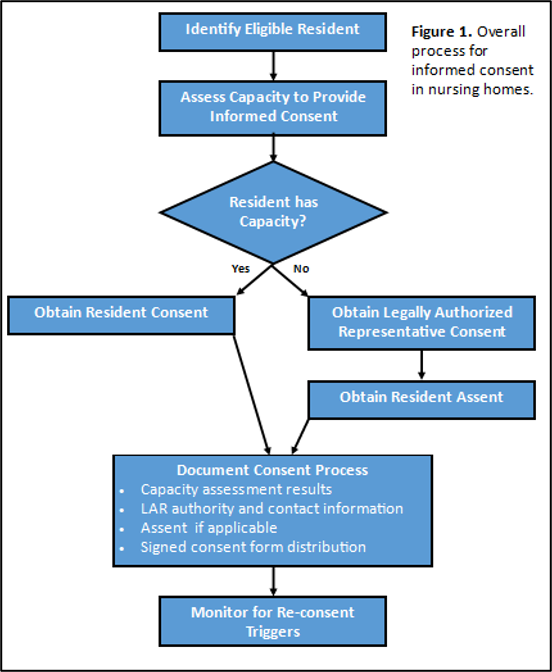
Practical Issues for Informed Consent in Nursing Homes
Who should obtain consent. Although NH staff are on site and have trusted relationships with residents and families that facilitate consent discussions, it is usually neither practical nor advisable for NH staff to lead the research consent process. First, having NH staff obtain consent meets the federal definition of the NH being “engaged in research,” triggering additional requirements for research ethics training, IRB oversight, and a Federalwide Assurance number. Second, competing clinical demands make the additional effort of consenting residents challenging for staff to fulfill and research may not progress as a result. Finally, vulnerable residents may feel coerced to participate when asked by staff whom they rely on for daily care. These issues are avoided when study staff lead the consent process under the supervision of the investigator, recognizing that this may increase the need for travel, impact the budget, and create additional logistical considerations. A blended approach can be used, in which the study is introduced by NH staff and a “warm handoff” is made to the study team to complete the informed consent process. Other strategies that leverage NH staff in the consent process are listed in Table 2.
Logistical issues. Beyond regulatory considerations, successful informed consent processes in NHs require planning around operational constraints within the setting. Table 2 outlines several logistical issues and potential approaches to address them.
Table 2: Logistical Issues Related to Informed Consent in NHs with Potential Approaches
| Issue | Potential Approaches |
|---|---|
| Participant Identification and Eligibility Screening | – Study staff are granted read-only access to NH electronic health records (EHR) or other data sources – Study staff review a list of names identified through the NH EHR with NH staff to screen out residents who may be ineligible due to conditions not listed in the EHR – NH staff provide a list of potentially eligible residents and/or family/LAR contact information; screening is completed during a study visit (in-person or telephone) by study staff – Some IRBs do not allow direct outreach from study teams (“cold calls”) – NH staff can approach the individual for permission before providing their name to the study team – Investigators can provide a letter for the NH to distribute to potential participants or their families/LARs with a mechanism to opt out of further contact (e.g., phone number, NH staff notification) |
| Protecting Personally Identifiable Information (PII) (requires a Data Use Agreement [DUA]) | – The NH designates a quiet private space to hold informed consent discussions – Study staff create a secure communication channel for NH and study staff to share PII needed for consent (e.g., encrypted email, secure fax, secure data upload) – Study staff obtain secure storage for hard copy documents containing PII, including during transport between the NH and investigator’s institution (locked, portable storage container) – Study staff specify what data elements need to be shared and where pre-consent data are to be stored (e.g., screening logs, clinical information on screen failures) |
| Handling Signed Consent Forms | – Study staff obtain access to a printer and/or scanner in the NH – Study staff determine the process for providing a copy of signed consent form to participants (e.g., hard copy left in resident room, mailed to family/LAR) or request waiver of this aspect of consent – Study staff determine if the NH wishes to keep a copy of the informed consent document or scan it into the EHR |
| Coordination between NH and Study Staff | – Both parties agree on timing of consent visits to minimize interruption of clinical care |
Accommodations to Facilitate Conversations about Informed Consent with NH Residents
Simple strategies to accommodate cognitive, motor, and sensory impairments common in NH residents can greatly facilitate the informed consent process. These include:
- Hearing protocol – reduce ambient noise, ensure hearing aids are in place and turned on, have a portable amplification device available
- Vision protocol – use high-contrast materials, large font, add additional lighting, ensure residents who wear glasses have these available, provide a magnification lens
- Cognitive protocol – develop a clear simple message describing the study and helping participants understand the purpose and their role. Provide a visual study summary with graphics to depict key study activities. Ask participants if they would like to include others during the discussion.
- Motor protocol – provide a portable lap desk for study documents, request a waiver of documentation when appropriate, create a process for documenting consent from residents who lack the physical ability to sign their name
- Allow extra time to complete informed consent. Plan for multiple visits when needed.
- Ask NH staff to help identify the optimal time to approach the resident about participation; avoid approaching the resident in the late afternoon or after tiring activities.
Tools and training for study teams to help them accommodate older persons with multimorbidity in all aspects of research, including the consent process, can be found at https://5tsframework.duke.edu/
Capacity Assessment and Assent
Assessing Capacity. Two-thirds of NH residents in the U.S. have a diagnosis of dementia, and so assessing capacity to provide research informed consent is a critical step. Dementia is under-documented in the medical record and sometimes not recognized by staff; therefore, it is recommended that investigators assess and document capacity in all potential participants who are alert and verbal regardless of their medical history or staff report.
As in clinical decision making, the capacity to consent for research is decision specific. Because the complexity and risk of projects vary, a resident may have the capacity to consent for some types of research but not others. Cognitive screening test scores (e.g., MoCA, SLUMS, MMSE, MiniCog) do not assess decision-making ability directly and should not be used as a substitute for assessing capacity because they can misclassify capacity for a significant proportion of residents. For example, a study of NH research participants in the UK showed that MMSE score predicted capacity for consent with an area under the curve (AUC) of 0.86, with an optimal cut-point score of 13-14. However, using this threshold misclassified 24% of residents as lacking capacity when they actually were capable of providing informed consent, and 21% as having capacity when they did not.(2) Therefore, it is recommended that investigators use validated research capacity assessment tools rather than cognitive screening tests to determine capacity to consent.
Research capacity assessment tools help researchers to assess four elements: 1) the ability to understand information about the study, 2) appreciate its relevance to them, 3) reason about choices by weighing the risks and benefits, and 4) express a choice. Tools are tailored to the specific protocol and begin with the researcher summarizing the key information about the study. Residents are then asked questions to assess their understanding and reasoning, and their responses are typically scored (e.g., inadequate understanding, partial recall/flawed logic, clear and accurate understanding). Most tools do not have a defined cutoff score but require investigators to use the results in each domain to inform their clinical judgement. In practice, however, it is common to pre-define a minimum score for proceeding with independent informed consent in the IRB protocol and study operating procedures for consistency and clarity (e.g., all items scored at least 4 out of 5).
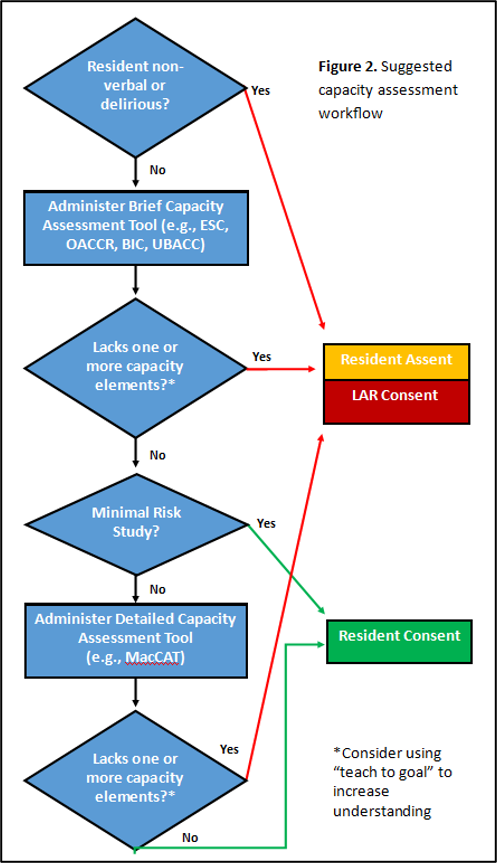
Tools vary in length and the training required to administer them, and there is no single tool that is appropriate for all NH research. Rather, the tool should be selected based on the study’s complexity and level of risk: brief tools may be sufficient for simple, low risk studies while significant risk studies should use a more detailed assessment (e.g., MacArthur Competence Assessment Tool for Clinical Research). In higher risk situations, a two-stage screening process can be employed, with a brief tool used to quickly identify those who clearly lack capacity, and the more comprehensive tool used to classify the rest. Select capacity assessment tools that have been validated in NH and/or cognitively impaired populations are listed below.(3)
Some investigators have successfully used a “teach to goal” consent strategy to increase the proportion who can consent on their own behalf.(12) After being read the consent information, potential participants are asked questions about the key elements of the study. For any elements that are answered incorrectly, the relevant section of the consent document is re-read or additional explanatory materials are provided. This process is repeated until all elements are answered correctly, or until a maximum number of attempts is reached. Teach to goal may be an especially useful strategy to improve generalizability in studies requiring active resident participation (e.g., advance care planning interventions) or when consent materials are complex. Investigators should describe this approach in their IRB protocol if they anticipate using iterative explanations to support participant understanding.
Assent for Residents Lacking Capacity. If a resident lacks the capacity to provide informed consent, investigators must still seek and document their assent to participate in research when feasible. Assent is defined as active agreement rather than simply failure to object to participation. Investigators should inform the resident about the research purpose, risks, and benefits in a way they can understand, then ask for their agreement to proceed. A resident’s verbal dissent or refusal must be respected, even if the family/LAR provided informed consent to participate. Depending on the type of study, assent may only need to be obtained once (e.g., one-time interview, application or wearable technology for prospective data collection), or before each study visit/procedure.
Sample script for requesting assent: “Hello, [name], I’d like to talk with you about a project I’m doing. We want to learn more about [simple description of study purpose in everyday language], and are asking if you’ll [simple description of study procedures]. Your [family member] said it’s okay for you to participate, but what matters most is how you feel about it. You don’t have to do anything you don’t want to do. Do you feel comfortable taking part? If at any time you want to stop, just tell me or show me and we will stop right away. Your comfort is important to me.”
Some residents may be incapable of providing verbal assent or dissent due to moderate to severe dementia or brain injury. Nonverbal signs that a resident is dissenting to participation include facial expressions, body posture, gestures or actions, or vocalizations suggesting distress. Investigators should work with their IRB to determine the appropriate process in the event that the residents are unable to provide verbal assent.
Suggested Capacity Assessment Workflow is found in Figure 2.
Table 3: Capacity for Research Consent Tools
| Capacity Assessment Tool | Description | Validation |
|---|---|---|
| MacArthur Competence Assessment Tool for Clinical Research (MacCAT-CR) (4) | Semi-structured interview format with 21 items, 20-30 minutes, assesses all domains of capacity | Considered a gold standard, validated in cognitively impaired and seriously ill populations (5, 6) |
| University of California San Diego Brief Assessment of Capacity to Consent (UBACC) (7) | 10 items, 5-7 minutes, assessing understanding, appreciation of voluntariness, and reasoning | Validated in mental illness, cognitive impairment (8) |
| Evaluation to Sign Consent (ESC) | 5 items, <5 minutes using a “teach back” format to assess understanding of study risks and procedures | Validated in NH residents with cognitive impairment (9) |
| Brief Informed Consent Test (BIC) | 11 True-False items, 5-10 minutes, primarily assessing understanding | Validated in older adults with dementia (10) |
| Older Adults’ Capacity to Consent to Research (OACCR) | 4 items, <5 minutes, assesses understanding and voluntariness | Validated in NH residents in Korea (11) |
Figure 2: Suggested Capacity Assessment Workflow
Informed Consent from Families/LARs
Family/LAR Perspectives on Research Participation. Studies suggest that families/LARs authorize participation at higher rates than patients would, especially in minimal risk studies.(13) A qualitative study of family/LAR decision-making for older adults with dementia who were approached to participate in a clinical trial suggested that reducing the burdens and risks of participation as much as possible, describing safety monitoring protocols, and obtaining the endorsement of the primary medical team were strategies that increased their willingness to consent for research for their loved one.(14)
Resident Representative vs. LAR. All NHs are required to list a resident representative in the health record –an individual chosen by the resident (generally a family member or friend) to assist with decision-making, access records, manage finances, and receive notifications on their behalf. In contrast, a LAR holds specific, formal legal authority to make decisions when a resident lacks capacity (e.g., guardian, power of attorney for healthcare, health care proxy). The resident representative may or may not also be the LAR, therefore, the investigator needs to verify and document who has decision-making authority for the resident. If a LAR is not clearly identified by the NH, the investigator should verify with the resident representative whether they are the legal guardian, power of attorney for healthcare, health care proxy, or (in the absence of these documents) the resident’s next of kin. Next of kin designations vary by state, and the investigator should ensure that they are familiar with relevant state law about the priority order and receive IRB approval for the process that will be used to identify the LAR.
Supporting Family/LAR Substituted Judgement. In most respects, the informed consent process is similar when obtained from a family member/LAR. The individual must receive the full informed consent discussion and documentation and must demonstrate that they understand the study well enough to make a decision on behalf of the resident. The main difference is that with standard consent, a resident uses their own preferences and values in decision making whereas a family member/LAR is ethically obligated to use substituted judgement (i.e., what the resident would have wanted), or if this is unknown, decide whether participation is in the resident’s best interest. It is advisable to include a reminder of this obligation in the consent script with a family member/LAR. It is also advisable to inform the family member/LAR that the team will obtain resident assent before starting any study procedures and describe how this will be done.
“Based on what you know about [resident’s] preferences and values, do you think [he/she] would want to participate in the research or not? If you are not sure, do you think it is it is [name’s] best interest to participate or not?”
“If you agree for [name] to participate, our team will tell [him/her] about the study’s purpose and make sure that [he/she] also agrees to participate before we proceed. If at any time [he/she] tells or shows us that [he/she] is uncomfortable, we will stop what we are doing.”
Logistical Considerations for Family/LAR Consent. Because many family members/LARs visit the NH infrequently, it is advisable to plan for informed consent discussions to occur over the phone or during a secure videoconference. Study teams may need to schedule informed consent meetings with the family member/LAR in advance and offer evening and weekend options to accommodate work schedules.
A waiver of documentation of informed consent may be appropriate for low-risk studies. If required, electronic signatures can be obtained from family members/LARs using REDCap, Adobe Sign, or other approved research tools, with a link sent via text or email to the family member/LAR. Copies of the signed informed consent document can be sent to the family member/LAR electronically. Some family members/LARs may lack digital capabilities, and having an IRB-approved option to mail written documents is advisable.
Re-Consent Plan
If the study poses significant ongoing risk or burden to participants over an extended time, investigators should plan for circumstances that may require re-consenting participants or their representatives.
Triggers for Reconsent. In addition to the usual requirement for re-consent based on new findings that change the risks/benefits of the study, the investigator should plan for reconsenting the resident and/or family/LAR during the study based on significant changes in the resident’s status. Reasons to consider re-consent include:
- Hospitalization or other serious acute illness
- Significant cognitive change in a resident who provided their own consent (e.g., new delirium, advancing dementia)
- Change in care goals (e.g., palliative care or hospice referral)
- Change in LAR (e.g., death of a spouse)
Depending on the level of risk and duration of the study, the investigator may need to develop a mechanism to proactively identify these events, such as a periodic review of the EHR or communication with NH staff. When identified, study staff should repeat the process of capacity assessment and, if indicated, pause study procedures until informed consent can be obtained from the current LAR.
NH Informed Consent Documentation Considerations
Study record documentation of the informed consent process with NH residents should include the following information:
- Results of the capacity assessment, and why the participant lacks capacity if applicable
- The identity, legal authority and full contact information of the family member/LAR, if applicable
- If family/LAR consent is obtained, participant’s assent or dissent
- Accommodations used to support understanding
- Confirmation that signed copies of the consent form were provided to the resident, family/LAR, and NH as applicable
- Need for re-consent for changes in participant capacity or change in family member/LAR during the study
- An impartial witness attesting to the completeness of the informed consent process is usually required if the resident/LAR is unable to read the consent form (due to blindness, literacy/language barriers, etc.) or is physically unable to sign it. IRB requirements for witness signatures vary, and investigators should seek guidance from their local experts.
Table 4: Best Practice Summary
| Element | Description | Best Practices for NH Research |
|---|---|---|
| Understanding | Information must be in simple, understandable language | – Accommodations for hearing, vision, and cognitive impairments available – Study staff training in communication with older adults |
| Voluntariness | Participation must be freely chosen, with the right to withdraw anytime | – Regular opportunities to opt out as goals change – Protocol flexibility to accommodate participant fatigue, availability (e.g., appointments, hospitalizations) |
| Competence | The individual must have the mental capacity to make the decision | – Capacity assessment process that includes the use of a validated research capacity assessment tool – Family/LAR identification and verification – Re-consent triggers for change in status |
| No Coercion | Avoid undue influence or pressure | – Study staff rather than NH staff obtain consent – Assent for individuals who lack capacity when feasible – Assurance that participation decision will not impact NH care provided |
| Opportunity to Ask Questions | Individuals must have time to ask questions and have them answered | – Offer second consent visit – Ask if participant wishes to include others during the informed consent discussion |
References
1. Sedrak MS, Ji J, Tiwari A, Mohile SG, Dale W, Le-Rademacher JG. Clinical Trial Enrollment, Ineligibility, and Reasons for Decline in Older vs Younger Patients With Cancer in the National Cancer Institute Community Oncology Research Program. JAMA Network Open. 2022;5(10):e2235714–e.
2. Whelan PJP, Oleszek J, Macdonald A, Gaughran F. The utility of the Mini-mental State Examination in guiding assessment of capacity to consent to research. International Psychogeriatrics. 2009;21(2):338–44.
3. Gilbert T, Bosquet A, Thomas-Antérion C, Bonnefoy M, Le Saux O. Assessing capacity to consent for research in cognitively impaired older patients. Clinical Interventions in Aging. 2017;12(null):1553–63.
4. Cutler B. Encyclopedia of Psychology and Law.
5. Goswami R, Moore J, Bruera E, Hui D. Assessment of the Decision-Making Capacity for Clinical Research Participation in Patients With Advanced Cancer in the Last Weeks of Life. J Pain Symptom Manage. 2020;60(2):400–6.
6. Palmer BW, Dunn LB, Appelbaum PS, Mudaliar S, Thal L, Henry R, et al. Assessment of Capacity to Consent to Research Among Older Persons With Schizophrenia, Alzheimer Disease, or Diabetes Mellitus: Comparison of a 3-Item Questionnaire With a Comprehensive Standardized Capacity Instrument. Archives of General Psychiatry. 2005;62(7):726–33.
7. Jeste DV, Palmer BW, Appelbaum PS, Golshan S, Glorioso D, Dunn LB, et al. A new brief instrument for assessing decisional capacity for clinical research. Arch Gen Psychiatry. 2007;64(8):966–74.
8. Duron E, Boulay M, Vidal J-S, El Bchiri J, Fraisse M-L, Rigaud AS, et al. Capacity to consent to biomedical research’s evaluation among older cognitively impaired patients. A study to validate the University of California Brief Assessment of Capacity to Consent questionnaire in French among older cognitively impaired patients. The Journal of nutrition, health and aging. 2013;17(4):385–9.
9. Resnick B, Gruber-Baldini AL, Pretzer-Aboff I, Galik E, Buie VC, Russ K, et al. Reliability and Validity of the Evaluation to Sign Consent Measure. The Gerontologist. 2007;47(1):69–77.
10. Buckles VD, Powlishta KK, Palmer JL, Coats M, Hosto T, Buckley A, et al. Understanding of informed consent by demented individuals. Neurology. 2003;61(12):1662–6.
11. Lee M. The Capacity to Consent to Research Among Older Adults. Educational Gerontology. 2010;36(7):592–603.
12. Sudore RL, Landefeld CS, Williams BA, Barnes DE, Lindquist K, Schillinger D. Use of a modified informed consent process among vulnerable patients: a descriptive study. J Gen Intern Med. 2006;21(8):867–73.
13. Berger JT, DeRenzo EG, Schwartz J. Surrogate decision making: reconciling ethical theory and clinical practice. Ann Intern Med. 2008;149(1):48–53.
14. Bogaerts JMK, Warmerdam LA, Achterberg WP, Gussekloo J, Poortvliet RKE. Proxy Decision-Making for Clinical Research in Nursing Home Residents with Dementia: A Qualitative Analysis. J Am Med Dir Assoc. 2023;24(4):541–7.e2.